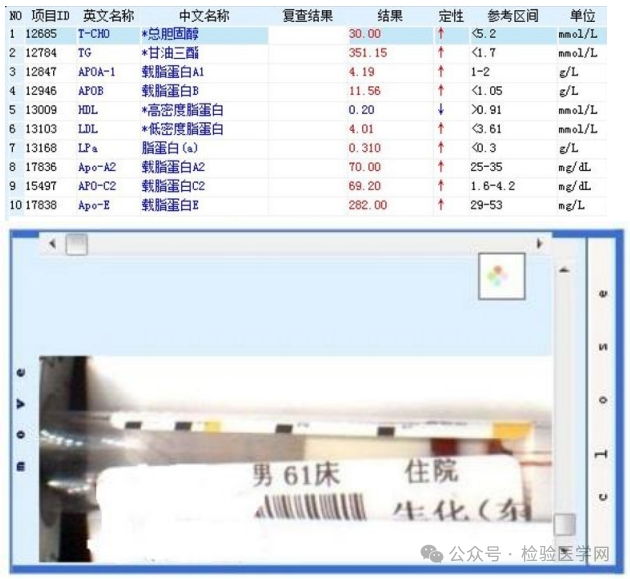
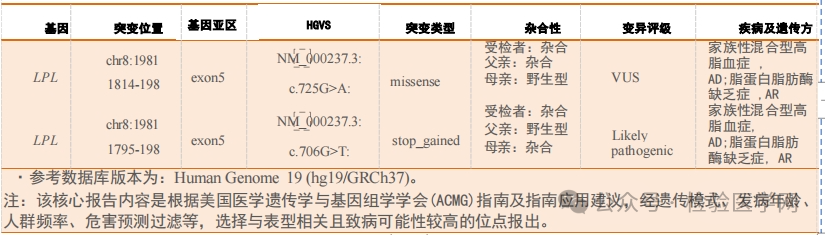
|
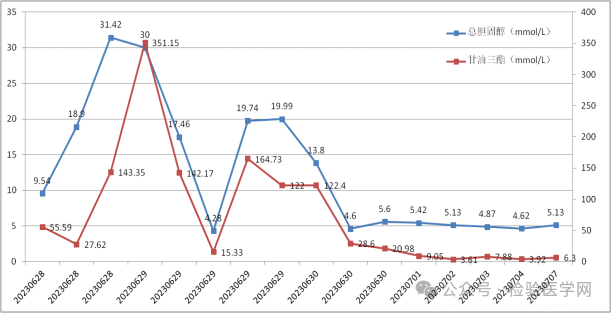
家族性混合型高脂血症(familial combined hyperlipidemia, FCHL)是冠心病患者中较常见的脂质代谢异常,是多基因遗传病。本文介绍一例罕见的婴儿高脂血症的临床特征、诊断和治疗经过,探秘婴儿高脂血症的发病机制,提醒家长关注婴儿高脂血症,守护宝宝健康成长。 一般资料 患儿男,1月,主因腹泻1天、便血半天、发现血液呈粉红色7小时入院。1天前患儿出现腹泻,为黄绿色、无臭味、有奶瓣,共4次。伴哄睡困难,进奶量减少,无呕吐、发热等情况,遂至当地诊所就诊,给予口服“斯唯诺口服液 3ml”1次。患儿仍腹泻,次数增多,且大便出现少量血丝,患儿家属未在意、未处理。半天前,患儿腹泻未缓解,大便血量增多,可见片状粉红色血,遂于当地人民医院NICU住院治疗,查肠管彩超:未见明显异常,排除肠套叠。患儿入院后急查血,发现血液为粉红色,建议转至上级医院就诊。 为求进一步诊治,患儿急就诊于我院,急诊以“便血、血液异常查因”收入院。发病以来,患儿神志清楚,精神稍差,食欲欠佳,睡眠增多,小便较少,体重无减轻,稍发热,查体:前囟 3cm×3cm、头围 39cm、身长 58cm、体重 5.65Kg,全身皮肤黏膜稍苍白,呼吸 32次/分,双肺听诊无异常,心率 123次/分,律齐,未闻及病理性杂音,腹部柔软,肠鸣音正常,肝肋下3cm可触及,质软,脾肋下 2cm,神经系统查体未见明显异常。 入院完善相关检查:粪便常规自动分析:隐血免疫法 +-;红细胞沉降率测定:血沉,严重脂血无法测出;急查血气分析:血糖 6.2mmol/L、乳酸 3.5mmol/L、血钠 135mmol/L,血钾、血钙、血氯正常,血红蛋白不能测出。由于患儿严重高血脂,多项检查结果未报告。 将采集的外周血经3500rpm离心5分钟后,可以观察到患者的血清呈现出牛奶样混浊的重度脂血。将脂血的血清用生理盐水分别稀释2倍、5倍和10倍,最终得出的患者第一次换血前后血脂结果如下图所示。 左右滑动查看更多 采血时发现患儿血液呈粉红色,放置后血浆呈乳白色,查血脂明显异常,甘油三酯极高(TG 351.15mmol/L ↑),考虑该患儿可能存在先天性脂代谢紊乱性疾病,应与以下疾病相鉴别: 是与该患儿症状较为相似的一种遗传性疾病。其与多种脂代谢相关的酶和脂蛋白基因缺陷相关,如脂蛋白脂肪酶(lipoprteinlipase, LPL)基因、胆固醇7-羟化酶基因、LPL激活剂载脂蛋白C-II (Apo CII)、Apo AII、Apo AI/C3/A4/A5基因簇和ApoE。 该患儿需要与一些罕见的先天性脂代谢紊乱性疾病相鉴别:
是与溶酶体酸性脂酶(lysosomal acid lipase, LAL)基因缺陷有关的常染色体隐性疾病。LAL在胆固醇酯和TG的水解中起重要作用,该病通常在 1 岁前发病,除伴有高 TG 血症和高 TCH 血症外,还出现肝脾大、脂泻、腹胀、肾上腺钙化和生长发育停滞等症状体征,并伴有多器官的脂质沉积 。
是常染色体隐性遗传性疾病,其发病原因为ABHD5基因突变导致其编码的蛋白质不能激活脂肪TG脂酶(Adipose triglyceride lipase, ATGL)。ATGL是将甘油三酸酯裂解成小分子结构为机体提供能量的重要物质,ATGL活性的下降,直接影响甘油三酸酯的分解,使TG类物质贮存在全身细胞内部。其主要临床表现包括皮肤鱼鳞病样改变、血液系统异常和共济失调等。
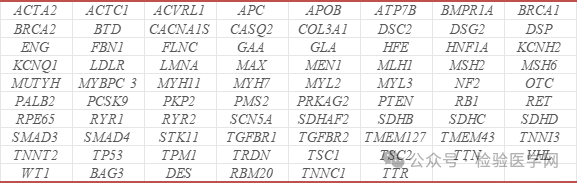
又称鱼眼病,是一种罕见的显性遗传性疾病,该病由于血浆磷脂酰胆碱-胆固醇酰基转移酶活性缺失,导致患者血浆TG和TC水平增高,并伴有蛋白尿、角膜混浊和贫血。较为特征的实验室检查,如脂蛋白电泳显示,高密度脂蛋白亚类(HDL3)组分缺乏而出现相对分子质量为115000的异常的HDL颗粒。 为了明确致病原因,我们采集了该患者及父母的外周血,使用全外显子组高通量测序检测技术,利用贝瑞基因自主研发的 Verita Trekker®变异位点检测系统和Enliven®变异位点注释解读系统对数据进行分析。经检测,ACMGSF v3.1(Miller et al., 2022)推荐报导的78个基因无异常。 Miller et al., 2022推荐的78个基因列表
[1] Yang L, Li Z, Song Y, et al. Study on urine metabolic profiling and pathogenesis of hyperlipidemia. Clinica Chim Acta, 2019, 495: 365-373. [2] Nagasaka H, Kikuta H, Chiba H, et al. Two cases with transient lipoprotein lipase( LPL)activity impairment: Evidence for the possible involvement of an LPL inhibitor [J]. Eur J Pediatr, 2003, 162(3):132-138. [3] GILBERT B, ROUIS M, GRIGLIO S, et al. Lipoprotein lipase (LPL) deficiency: a new patient homozygote for the preponderant mutation Gly188Glu in the human LPL gene and review of reported mutations: 75% are clustered in exons 5 and 6[J]. Ann Genet, 2001, 44(1): 25-32. DOI: 10.1016/s0003-3995(01)01037-1 . [4] H, Knoblauch, A,Busjahn, S, Münter, Z, Nagy, HD,Faulhaber, H,Schuster, F C,Luft. Heritability analysis of lipids and three gene loci in twins link the macrophage scavenger receptor to HDL cholesterol concentrations.[J]. Arteriosclerosis, thrombosis, and vascular biology, 1997, 17(10):2054-60.DOI:10.1161/01.atv.17.10.2054. [5] 田时秋, 李依林, 裴海鸾, 田颖颖, 左泽平, 赵新月, 刘闯, 王志斌.高脂血症发病机制及药物治疗[J].生命的化学,2022,42(12):2237-2247. |
Copyright © 2015-2023 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号