|
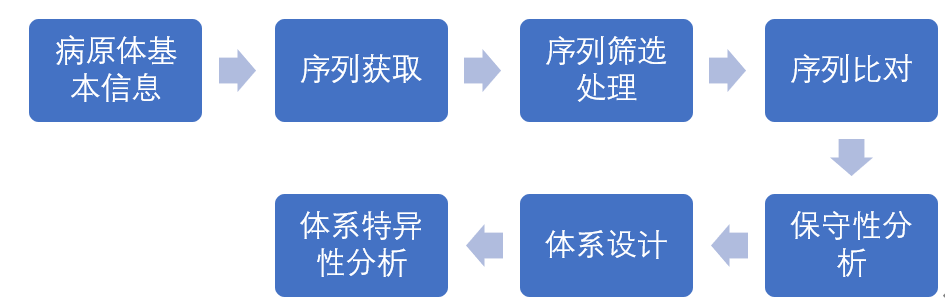
最近大家都想了解多重PCR检测病原体的体系设计方法,剽了赵博的经验,整理出了一份基于多重PCR技术的病原体体系设计指导原则,文档比较长,干货比较多,适合专业人士学习,小白看看就行了!!! 目录如下,嫌弃长的赶紧绕道,别怪没提醒!!! 目录: 1 体系设计流程... 2 2 病原体信息获取... 2 3 序列信息获取... 2 4 序列处理筛选... 3 5 序列比对... 3 5.1 MEGA11使用方式... 3 5.2 MAFFT安装及使用说明... 4 6 保守性分析... 5 6.1 保守性分析... 5 6.2 寻找保守序列... 6 7 体系设计... 7 7.1 Primer
Blast引物设计方法... 8 7.2 根据保守性手动设计引物探针... 8 8 体系特异性分析... 9 1 体系设计流程
2 病原体信息获取信息来源文献报道、专利等公开资料。需要获取的信息如下:病原体中英文名称、别名等,及现有检测的常用靶点名称等信息; 基因组核酸种类(长度、结构、DNA/RNA、单链/双链、正链/负链等信息); 确认是否存在分型,及具体分型名称及分型原则(例如流感病毒分为甲流、乙流、丙流等;甲流根据H基因和N基因分为H1N1/H7N9; 确认是否存在近源物种,并确认近源物种的相关信息及交叉反应的风险(如甲流通常感染禽类,但有一部分能感染人;冠状病毒来源于牛、蝙蝠等动物,少部分能感染人);
3 序列信息获取从NCBI基因组数据库或其它公开数据库获取序列信息,常用数据库:
NCBI基因组数据库,https://www.ncbi.nlm.nih.gov/genome/ NCBI核酸数据库,https://www.ncbi.nlm.nih.gov/nucleotide/ NCBI病毒数据库,https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/
序列下载原则: 尽可能多的获取基因序列; 优先获取完整基因组序列; 优先下载宿主为人的基因序列;
4 序列处理筛选如序列信息较多,则需要进行筛选,常用筛选流程有: 5 序列比对使用Mega、Mafft等软件进行分析,序列比对的目的是为后续保守性分析做准备。 序列较少时使用Mega进行分析(500条以内), 序列较多时需要使用Mafft进行分析;
5.1 MEGA11使用方式
- 所用序列需要为FASTA格式,使用MEGA11打开处理后的病原体核酸序列;
- 选择全部序列,并使用MUSCLE算法进行序列比对,参数为默认即可;
- 保存MEGA比对文件,并将比对后的序列保存为FASTA格式用于后续分析,文件名称为“病原体代号_Align.fa”;
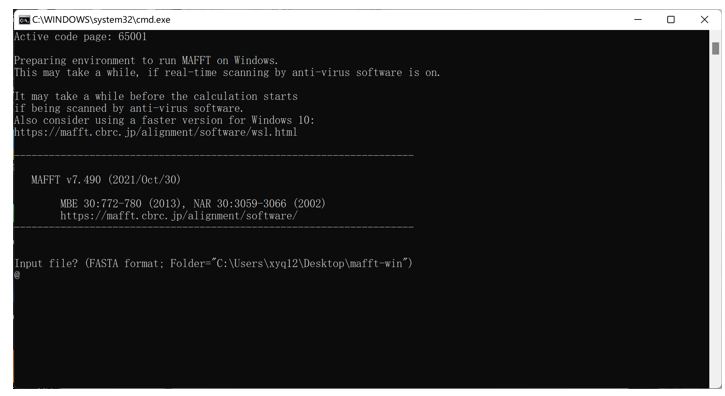
5.2 MAFFT安装及使用说明安装及使用方式可参考以下链接:https://mafft.cbrc.jp/alignment/software/windows_without_cygwin.html,具体如下: 下载MAFFT-Windows版本安装包,并解压; 将待比对文件复制到解压后的文件夹中; 单击MAFFT.bat,输入待比对文件名称(文件名包括文件后缀,如test.fa); 确认输出的文件名称、输出顺序(建议为4)、其它参数(为空即可),并输入“Y”确认开始比对;
对完成后的序列文件可直接用于后续分析或处理。
6 保守性分析 6.1 保守性分析先人工判断比对后序列的保守性,如保守性较好则可直接进行后续设计,如保守性较差,则使用GeneFisher找出95%或99%的保守区域。GeneFisher网站,https://bibiserv.cebitec.uni-bielefeld.de/genefisher2/。具体使用方式如下:该网站输入序列为比对后的FASTA文件,所有文件需要有相同的长度(空碱基用“-”表示); 根据以上链接打开genefisher2网站,选择Submission开始上传序列文件,模式选择第三项“c)DNA”,并单击next进入文件上传页面; 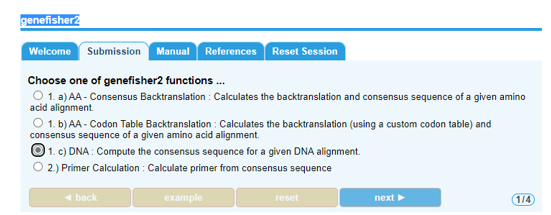
选择需要上传的序列文件,并单击next进行阈值线设置页面。再次提醒,上传文件需要为比对后的FASTA格式文件,如存在序列长度不一致的文件会导致分析失败; 需要尝试多种阈值线进行保守型分析,以确认最佳的保守区域,建议先选择99/98/95三个阈值进行分析; 
确认阈值线后开始进行保守性计算,并保留所有阈值线下的分析结果,并根据阈值线进行标记。分析结果可直接下载或以文本方式显示; 
6.2 寻找保守序列使用MEGA打开比对后的序列文件,并将分析得到的保守序列复制到其中;下图方框内即为分析得到的保守序列,并根据阈值线进行命名;需要综合考虑不同阈值线下的保守序列,并优先选择最为保守的区域,确保把最保守的区域留给探针。 
然后,剩下的交给运气了!!GOOD LUCK!! 7 体系设计使用SnapGene保存引物探针序列,引物探针设计原则如下: - 如序列保守性较高,则可考虑使用NCBI Primer Blast设计引物;
- 扩增片段长度不超过150bp,通常在100bp左右;
- 引物Tm值在60℃左右(使用IDT分析,在3mM镁离子、50mM钠离子、0.8mM
dNTP和0.2μM Oligo的条件下计算Tm值),https://sg.idtdna.com/calc/analyzer
- 对于RNA病毒,探针应设计在非反转录方向上(不干扰逆转录过程)
- 根据序列特意引物Tm值可适当提高,通常不超过65℃都可以接受;
- 对于保守性极高的序列(突变概率低于0.1%,拥有连续200bp以上的完全保守序列),使用Primer Blast根据特异性设计引物组合;在线工具地址链接:https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome。
- 打开以上地址,输入序列信息(不超过50000bp)。
- 引物设计参数:扩增片段长度70-150bp,Tm值55-60℃,最适Tm值为57℃;
- 特异性参比数据库选择人基因组、全部细菌基因组和全部病毒基因组;

7.2 根据保守性手动设计引物探针
- 如果序列保守性较高或序列较长,则优先根据文献或专利等信息确认常用检测基因或片段范围;
- 如保守性较差(保守序列较少)或基因序列较短,则在全基因组范围内寻找合适的检测片段;目前尝试过10Kb基因组范围内寻找保守片段(HIV);
- 从NCBI GenBank上下载一条完整的病原体基因组序列(参考序列),建议序列格式为.gb文件,并用Snapgene打开;
- 将长度超过18bp且至多只有一个简并碱基的99%保守度的序列全部以引物的形式添加至此序列中;(可接受简并碱基数量可根据序列保守程度调整);
- 将最为保守的区域作为探针序列,并寻找扩增长度可以在70-100bp左右(150bp以内)的引物探针组合;
- 上下游引物若没有合适的99%保守度的序列则可以选择95%或98%保守度序列。
8 体系特异性分析使用NCBI Primer Blast对引物进行特异性分析。将设计的引物复制至Primer
Blast,对比数据库为参比数据库选择人基因组(Homo Sapiens)、全部病毒基因组(Viruses)和全部细菌基因组(Bacteria);限制扩增片段长度为300bp。使用MFE Primer 3.1在线工具(https://mfeprimer3.igenetech.com/)对所有设计的引物探针二聚体分析和二级结构分析,二聚体分析参数如下:允许的错配阈值为5、同时显示3‘和5‘端形成的二聚体结构。 恭喜你,是不是很多都软件都不会用,不要着急,慢慢来哈!!!
|



