|
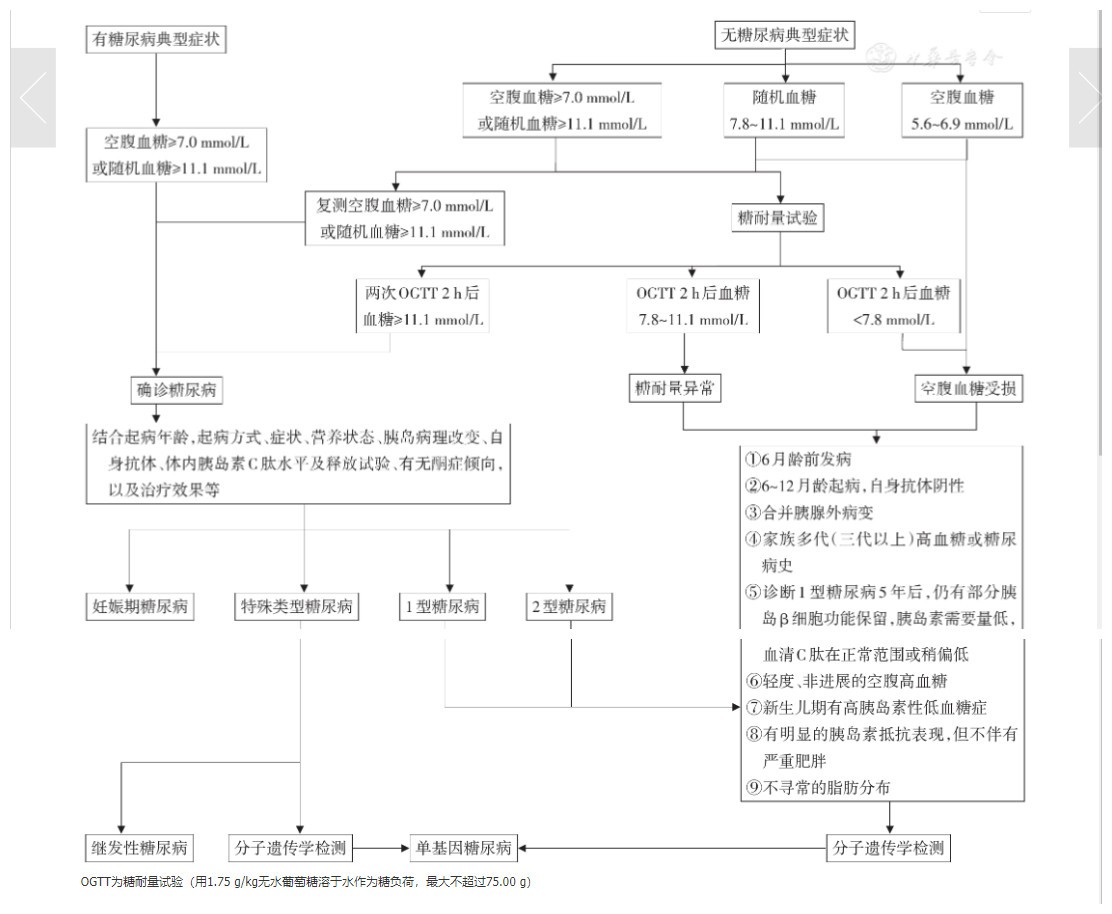
摘要 随着对糖尿病发病机制认识的深入,以单基因糖尿病为主的特殊类型糖尿病正日益引起人们的关注。单基因糖尿病变异占儿童糖尿病的1%~6%,但大多数单基因糖尿病在早期易被误诊。分子遗传学检测是确诊单基因糖尿病的首要方法,为进一步规范中国儿童单基因糖尿病的诊断、分型、治疗、随访、并发症和合并症的评估及防治,由中华医学会儿科学分会内分泌遗传代谢学组组织专家讨论,并参考国内外的最新指南,同时结合国内外流行病学调查、临床研究成果,制订本共识。 糖尿病是一个日益严重的全球公共卫生问题,据2017年国际糖尿病联盟报道,糖尿病患病总数及经济支出逐年上升,有30%~80%的糖尿病患儿没有及时得到确诊和正确的分型,导致并发症及病死率上升。2017年中国儿童糖尿病总患病数已达4.7万人,每年新增患病数0.6万人,均位居全球排名第4[1],严重影响我国儿童的健康成长。 根据1999年世界卫生组织提出的标准,糖尿病分为1型糖尿病(type 1 diabetes mellitus,T1DM)、2型糖尿病(type 2 diabetes mellitus,T2DM)、妊娠期糖尿病和特殊类型糖尿病。单基因糖尿病占儿童糖尿病的1%~6%,但大多数单基因糖尿病在早期易被误诊[2],为进一步规范中国儿童青少年单基因糖尿病的诊断、分型、治疗、随访、并发症和合并症的评估及防治,由中华医学会儿科学分会内分泌遗传代谢学组组织专家讨论,并参考国内外的最新指南,同时结合国内外流行病学调查、临床研究成果,制订本共识。 (一)NDM NDM多指6月龄以内发病的糖尿病,已知超过30种单基因相关NDM的遗传亚型。按病程可分为持续性新生儿糖尿病(persistent neonatal diabetes mellitus,PNDM)和暂时性新生儿糖尿病(transient neonatal diabetes mellitus,TNDM),合并胰腺外病变者多为综合征型。TNDM者的糖尿病症状会在几周或几个月内缓解,但在儿童期或之后可能复发[3]。所有6月龄前发病的糖尿病患儿都应进行基因检测,早期的基因检测将有助于分型和判定预后。常见的NDM基因变异如下。 1.6q24印记区域异常导致TNDM: 60%以上的TNDM是由染色体6q24印记区域的基因变异或甲基化异常引起的,以PLAGL1基因和葡萄胎相关的印记转录子-1基因(HYMAI)变异最为常见[4]。TNDM发病与印记基因的过度表达有关,有3种分子机制,(1)6号染色体的父系单亲二倍体,占散发性TNDM的50%,也可导致PNDM[5],同胞和后代的发病风险很低;(2)6q24区域的父系重复,多为家族性,同胞和后代50%的新生男性可发病或携带,女性可见隔代遗传;(3)母亲等位基因的异常甲基化,多为散发。临床多表现为严重的宫内发育迟缓,出生后1周内出现严重的、非酮症性高血糖(平均诊断年龄0~1周龄)。1/3的患儿可合并巨舌,也可出现脐疝、先天性心脏缺陷、脑畸形等临床特征。治疗上,对于多数TNDM患儿,胰岛素治疗后剂量可逐渐减停,平均缓解年龄13周龄,观察期为18周[6]。有50%~60%的患儿会在青春期前后复发,已知最小复发年龄为4岁[7]。复发患儿大多数仍保留一定程度的胰岛β细胞功能,一般不需胰岛素治疗,口服磺脲类药物可有一定治疗效果[8,9]。值得注意的是,少部分患儿在缓解后会出现低血糖,严重者需要长期治疗。 2.ATP敏感性钾通道(ATP sensitive potassium channel,KATP)基因变异: KATP是由4个成孔Kir6.2亚单位和4个SUR1调节亚单位形成的异八聚体复合物,分别由KCNJ11和ABCC8基因编码,多为杂合变异,可阻止钾离子通道关闭、抑制胰岛素分泌,导致高血糖,是引起PNDM的最常见致病基因,也是导致TNDM的第二大原因。KCNJ11变异多导致PNDM,ABCC8变异多导致TNDM。家族病例中呈现常染色体显性遗传,后代发病风险为50%。部分纯合或复合杂合变异,为隐性遗传,同胞发病风险为25%,后代多不发病。临床易出现糖尿病酮症酸中毒[10],血清胰岛素及C肽水平低。TNDM者多有轻度宫内发育迟缓,发病和缓解年龄均迟于6q24-TNDM,平均诊断年龄为4周龄,平均缓解年龄为35周龄,但复发时间较早。由于在神经和肌肉组织中同样表达KATP,除糖尿病表现外,所有KATP基因变异患儿均存在轻度神经系统发育异常,如发育性共济障碍(特别是视觉空间障碍)、注意力缺陷多动障碍、执行功能下降、焦虑症或自闭症、睡眠障碍等[11]。约20%的KCNJ11变异患儿可出现严重神经系统发育异常,表现为发育迟缓、早发癫痫,称为NDM发育迟缓癫痫综合征,其余大部分表现为NDM、轻度发育迟缓且不伴癫痫的中度NDM伴轻度发育迟缓癫痫综合征。相比而言,ABCC8变异患儿的神经系统并发症轻微且罕见。90%以上可使用较高剂量磺脲类药物替代胰岛素治疗,以格列本脲为例,平均治疗剂量0.5 mg/(kg·d),最大可达2.3 mg/(kg·d)[6,9],血糖控制后磺脲类药物可逐渐减量。1岁前开始用药的患儿视觉运动和视觉感知功能较其他患儿可有明显改善,但仍明显落后于正常患儿[12]。临床上应密切关注高剂量磺脲类药物引起的严重低血糖反应、胃肠道反应、过敏反应及肝肾功能损害,及时减量或者停药。用药前需取得监护人的知情同意。 3.INS基因变异导致的NDM: INS基因杂合变异是PNDM的第二大常见原因。变异导致胰岛素原分子的错误折叠并聚集于内质网,引起内质网应激和β细胞凋亡。多为散发,约20%具有常染色体显性遗传的NDM家族史,罕见纯合或复合杂合变异。临床表现上,宫内发育迟缓的程度与KATP-PNDM相似,但发病时间较晚,可在6月龄后发病,胰岛素水平偏低,多不伴随神经系统异常表现。治疗上,发病后需终身使用外源性胰岛素控制血糖[6],胰岛素方案因个体化,起始剂量可同T1DM,也有少数案例应用磺脲类药物替代胰岛素治疗,但尚需进一步临床验证[9]。 4.Wolcott-Rallison综合征: 由EIF2AK3基因纯合变异或复合杂合变异引起的罕见常染色体隐性遗传综合征。EIF2AK3编码蛋白调节内质网应激反应,错误折叠的蛋白质在内质网中积累并最终诱导β细胞凋亡。常染色体隐性遗传,父母为近亲结婚的患儿,尽早进行基因检测。同胞发病风险为25%。临床表现为PNDM、脊柱发育不良、多发性骨骺发育不良、复发性肝功能肾功能不全。婴儿期血糖增高多为其首发临床表现,其他症状可能到3~4岁才出现。治疗上,血糖控制依赖外源性胰岛素治疗,但血糖控制效果多不佳。并发症骨折需矫形治疗,肝肾功能不全需积极对症治疗,该病患儿易发生多器官功能衰竭,预后不佳。 5.IPEX综合征: IPEX综合征由FOXP3基因变异所致,是唯一确认与β细胞自身免疫和胰岛自身抗体相关的PNDM。FOXP3位于Xp11.23,参与调节T细胞发育、抑制自身免疫,其缺陷导致X连锁隐性遗传的自身免疫功能缺陷性多内分泌腺体病和肠病,男性多见,女性携带者多无临床表现。临床表现除PNDM外,多有新生儿腹泻、湿疹、自身免疫性甲状腺疾病,易出现危及生命的感染。治疗上,血糖控制需依赖胰岛素治疗,可使用免疫抑制剂西罗莫司或类固醇激素治疗,同种异体骨髓干细胞移植可能有效。 (二)MODY MODY是以胰岛素分泌受损为特征的单基因糖尿病,患儿多保留一定的胰岛素分泌能力,呈常染色体显性遗传。如无家族史提供,易被误诊为T1DM或T2DM。已报道14种不同的基因变异引起MODY,部分基因变异可引起除糖尿病外的其他临床特征,例如肾囊肿或胰腺外分泌功能障碍。MODY的不同遗传亚型在发病年龄、高血糖情况和治疗方案各有不同[13]。最常见的3个致病基因为HNF4A(maturity-onset diabetes of the young 1,MODY1)、GCK(maturity-onset diabetes of the young 2,MODY2)、和HNF1A(maturity-onset diabetes of the young 3,MODY3)。 1.HNF4A-MODY(MODY1型): HNF4A基因杂合变异导致。HNF4A位于20q13.12,其编码的蛋白在胚胎发育早期表达于胰腺胚芽PDX-1阳性细胞中,晚期广泛表达于所有胰腺内分泌细胞、外分泌细胞。HNF4A基因缺陷可导致双重临床表现,15%出生时可发生对二氮嗪敏感的高胰岛素血症性低血糖,青少年时期可表现出糖耐量异常,后期逐渐出现空腹血糖升高和糖尿病的"三多一少"症状,但很少出现酮症。临床还可见巨大儿,骨软化、氨基酸尿、高磷酸尿及血脂代谢紊乱等表现[14]。治疗上,大部分对磺脲类药物治疗敏感,但应注意,儿童应从低剂量起用(成人正常起始剂量的1/4),用药前需取得监护人的知情同意,并密切关注其用药安全性及不良反应如严重的低血糖、胃肠道反应、过敏反应及肝肾功能损伤等。磺脲类药物控制不佳或不能耐受者需胰岛素治疗。 2.GCK-MODY(MODY2型): 葡萄糖激酶是胰岛β细胞的葡萄糖感受器,是葡萄糖磷酸化步骤中的关键限速酶。由GCK基因杂合变异引起的MODY2型,是无症状家族性高血糖最常见的病因,呈常染色体显性遗传,MODY2型患儿调节胰岛素分泌功能多正常,仅调节血糖的阈值偏高。GCK纯合变异或复合杂合变异可导致PNDM,呈常染色体隐性遗传,同胞发病风险较高为25%。临床表现为家族性、轻度、非进展性高血糖(5.5~8.0 mmol/L),常无糖尿病经典"三多一少"症状,多在体检时发现,早期无明显糖耐量异常,到青春期或成年期逐渐出现糖耐量异常,餐后血糖或糖耐量试验2 h血糖较空腹可有显著增高(>5 mmol/L)[15]。而GCK基因纯合变异或复合杂合变异导致的PNDM,常在出生后几天即确诊为糖尿病,伴严重的宫内发育迟缓,一般无其他胰腺外病变表现。治疗上,对于MODY2型,常规剂量的胰岛素或口服降糖药物并不能有效降低血糖或HbA1c,大多数可通过单纯饮食、运动来控制血糖,很少会出现远期微血管或大血管并发症[16]。而GCK基因纯合变异或复合杂合变异PNDM患儿,因完全性GCK基因缺陷导致高血糖环境下β细胞无法分泌胰岛素,多需外源性胰岛素治疗。 3.HNF1A-MODY(MODY3): 由HNF1A基因变异导致有症状的家族性单基因糖尿病中最常见的亚型,发病率约是HNF4A-MODY的10倍。其诊断年龄取决于变异发生的位置,母系遗传者由于宫内环境的影响将使患儿发病年龄提前约12年。HNF1A基因变异造成肾小管葡萄糖转运受损,会影响葡萄糖重吸,因此餐后尿糖阳性多先于临床高血糖前出现,临床表现为肾性糖尿。糖耐量异常多在青少年期出现,餐后或糖耐量试验2 h血糖显著升高,早期空腹血糖可正常,随年龄增长逐渐出现空腹血糖升高和糖尿病的"三多一少"症状,但很少出现酮症。HNF1A变异也可引起高胰岛素血症低血糖,但非常罕见。治疗上,可使用磺脲类药物代替胰岛素治疗来控制血糖。但必须注意磺脲类药物在儿童患儿中的用药安全性,尽量从低剂量(成人正常起始剂量的1/4)开始,以避免发生低血糖。在不发生低血糖的情况下,可以长期维持低剂量的磺脲类药物。如果发生低血糖,可考虑使用该类药物的缓释剂或进餐时服用短效药物[17]。用药前需取得监护人的知情同意,并需关注该药的其他不良反应,如胃肠道反应、过敏反应及肝肾功能损伤等。HNF1A基因变异患儿心血管疾病和视网膜病变风险可增加,其他慢性并发症的出现与代谢控制水平相关。 4.HNF1B-MODY(maturity-onest diabetes of the yaung 4,MODY4型): 又被称作肾囊肿和糖尿病综合征。HNF1B基因杂合变异很少导致单纯的糖尿病症状,绝大部分都伴随肾囊肿或其他肾发育不良,可伴一定程度的肝源性胰岛素抵抗,因为1/3~2/3患儿为新发变异。临床表现以糖尿病、肾脏发育异常、女性泌尿生殖道畸形(特别是子宫畸形)为特征性表现,还可能出现高尿酸血症、痛风和肝肾功能异常。糖尿病症状出现较晚,通常发生在青春期或成年早期。胰腺外分泌功能下降,粪便弹性蛋白酶减少。胰腺影像学可见胰体和(或)胰尾部的缺失。治疗上,对磺脲类药物治疗无反应,需要早期胰岛素治疗,胰岛素方案应个体化。 5.MODY-胰腺外分泌综合征: 编码胰脂肪酶的CEL基因杂合变异,可导致胰腺外分泌功能不全和糖尿病,呈常染色体显性遗传。胰腺外分泌功能不全可早于糖尿病症状10~30年出现,故儿童期和青少年期诊断患儿极为罕见,粪弹性蛋白酶降低和胰腺脂肪瘤病可协助早期诊断。可伴有神经系统发育异常,以神经脱髓鞘病变多见。治疗上,血糖控制需依赖胰岛素治疗。伴有胰腺外分泌功能障碍者,还需补充胰酶。需要注意的是,其他单基因疾病如囊性纤维化(CFTR基因变异)、遗传性胰腺炎(PRSS1和SPINK1基因变异)和胰腺发育不全或先天性心脏病(GATA6基因变异)等常染色体显性遗传病也可影响胰腺外分泌,最终继发糖尿病,应注意根据临床特征及基因诊断结果鉴别。 (三)与糖尿病相关的其他遗传综合征 任何伴有多系统胰腺外病变的儿童糖尿病患儿都应考虑单基因糖尿病可能。 1.尿崩症、糖尿病、视神经萎缩和耳聋综合征(Wolfram综合征): 90%以上的患儿为隐性遗传的WFS1基因纯合变异,另一个罕见致病基因CISD2变异可引起出血倾向和消化性溃疡,但多无尿崩症表现。非自身免疫性的糖尿病通常是该疾病的首发临床表现,婴儿早期也可出现。进行性视神经萎缩引起的视力下降多在糖尿病诊断后数年发生,易被误诊为糖尿病视网膜病变。其他典型的临床特征,例如感应神经性耳聋、中枢性尿崩症、尿路功能障碍和神经系统等症状,出现顺序没有规律性。治疗上,发病后需终身依赖外源性胰岛素治疗控制血糖,胰岛素方案应个体化,起始剂量可同T1DM。醋酸去氨加压素改善多尿症状,配戴助听器改善耳聋等。 2.线粒体糖尿病: 由于线粒体变异和缺失导致的糖尿病在儿童中很少见(<1%)[18],绝大多数患儿在成年期发病。最常见的线粒体糖尿病是由线粒体DNA中的m.3243A>G变异引起的,其他也可见于如Kearns-Sayre综合征和Pearson综合征。线粒体糖尿病多呈母系遗传。起病较隐匿,20%的患儿可急性起病,并出现糖尿病酮症酸中毒,可合并感音神经性耳聋、黄斑病变和进行性外眼肌麻痹,可能导致肌肉疼痛、胃肠道症状、肾病、心肌病和神经精神症状。治疗上,早期可通过饮食运动控制或口服降糖药物治疗,起病数月或数年后即需依赖胰岛素。需要注意的是,二甲双胍可干扰线粒体功能诱发乳酸酸中毒,应避免使用。 3.单基因胰岛素抵抗综合征: 临床特征主要为高胰岛素血症、高胰岛素治疗量需求和中重度黑棘皮病,多不伴随严重肥胖,卵巢来源的高雄激素血症通常早于高血糖出现[19]。发病机制包括原发性胰岛素信号通路缺陷、继发于脂肪组织异常的胰岛素抵抗、以胰岛素抵抗为特征表现的复杂临床综合征。(1)由胰岛素受体基因(INSR)变异引起原发性胰岛素信号缺陷,导致多种罕见的胰岛素抵抗综合征。瘦素水平低,脂联素水平正常或升高。最常见的形式是A型胰岛素抵抗综合征,青少年期发病,女性多于男性,常染色体显性或隐性遗传,临床主要表现为与肥胖程度不符合的严重黑棘皮病和高雄激素血症。INSR基因纯合变异可导致更严重的Donohue综合征和Rabson-Mendenhall综合征,新生儿期及婴儿期即可见宫内发育不良,生长迟缓、黑棘皮病、多毛、性早熟、外生殖器肥大、餐后高血糖合并空腹低血糖。治疗上,可尝试胰岛素增敏剂,但大多数患儿需要高剂量胰岛素治疗,且效果有限,糖尿病远期并发症多见。对于儿童,重组人胰岛素样生长因子1或可改善空腹和餐后血糖,但对生存的长期影响尚不明确[20]。(2)单基因脂肪营养不良,临床特征为选择性四肢脂肪组织缺乏,下躯干和颈部周围皮下脂肪堆积,偶有软组织假性肢端肥大表现。内脏脂肪大大增加,肝脏脂肪变性、腹部膨隆和胰岛素抵抗。80%的先天性全身性脂肪代谢障碍(Berardinelli-Seip综合征)由AGPAT2基因或BSCL基因纯合变异导致,多为常染色体隐性遗传。LMNA基因和PPARG基因杂合变异也可引起该病。实验室检查除高胰岛素血症,高甘油三酯血症和低高密度脂蛋白胆固醇外,还有高雄激素血症的表现。治疗上,低脂肪、低热量饮食为主要控制手段。胰岛素增敏剂如二甲双胍和格列酮类可能有效,用药前需取得监护人的知情同意,并关注该药的其他不良反应,如格列酮类可引起面部和颈部脂肪的进一步积聚[19]、水肿、贫血及心功能不全等。严重先天性脂肪代谢障碍的患儿,可考虑重组瘦素治疗,可改善高甘油三酯血症、高血糖和肝脏脂肪堆积情况[21]。(3)纤毛病变相关的胰岛素抵抗和单基因糖尿病,Alström综合征由未知功能的ALMS1基因变异引起,多为常染色体隐性遗传。临床表现包括肥胖、黑棘皮病、高脂血症、高尿酸血症、高血压和慢性进行性胰岛素抵抗的糖尿病症状、视锥-视杆细胞退化、感觉神经性听力丧失等,超过60%的患儿合并心肌病。早期生活方式干预可以改善代谢异常[22]。Bardet-Biedl综合征以智力障碍为特征表现,同时有视锥-视杆细胞退化引起的进行性视力损害、多指畸形、肥胖、糖尿病、肾发育不良、肝纤维化和性腺机能减退。Bardet-Biedl综合征与21个位于不同染色体的基因变异相关联,称为BBS1至BBS21[23],多为常染色体隐性遗传。预后较差,肾功能衰竭是其主要死亡原因。 参考文献(略)
|
Copyright © 2015-2023 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号